Bài 16. Bệnh Học Nhiễm Sắc Thể Người
Tác nhân đột biến NST & Dấu hiệu lâm sàng
Vật lý
Sốc nhiệt độ cao (thấp) → Ức chế: Quá trình giảm phân → Giao tử 2n. Các lần phân bào đầu tiên của hợp tử. → Các tế bào đa bội.
Tia bức xạ ion hóa: tia UV, tia X và tia gamma. Tần số đột biến tỷ lệ: Liều lượng phóng xạ (không có liều vô hại), thời gian tiếp xúc và trạng thái cơ thể.
Hóa chất
Chống ung thư → Kìm hãm quá trình phân bào: Colchicine, Vinblastine, Vincristine, Taxol.
Vừa gây ung thư vừa gây đột biến: Chì, Benzen, thủy ngân, thuốc trừ sâu, diệt cỏ, …
Vai trò của Virut
Các Virut gây bệnh: Đậu mùa, Thủy đậu, Rubeola, Viên gan … → Bất thường cấu trúc NST.
Các dấu hiệu lâm sàng để phân tích NST
- Lịch sử gia đình
- Vấn đề sinh sản
- Phụ nữ mang thai trên 35 tuổi
- Thai chết lưu và tử vong sơ sinh
- Ung thư
- Các vấn đề tăng trưởng và phát triển trong gđ sớm
Danh pháp NST
A....G: Nhóm của các nhiễm sắc thể
1.....22: Các cặp nhiễm sắc thể
X và Y: Nhiễm sắc thể giới tính
I: Sự phân tách các dòng tế bào trong một cơ thể (thể khảm)
p: Nhánh ngắn của nhiễm sắc thể (petit)
q: Nhánh dài của nhiễm sắc thể (queue)
ptel (pter): Đầu tận cùng ở nhánh ngắn. (tel: telomeres; ter: terminal)
qtel (qter): Đầu tận cùng ở nhánh dài
+: Thừa nhiễm sắc thể
–: Thiếu nhiễm sắc thể
: Chỗ đứt
:: Đứt – Nối lại
mar: Nhiễm sắc thể đánh dấu (mark)
mat: Nguồn gốc từ mẹ (maternal)
pat: Nguồn gốc từ bố (paternal)
der: Xuất phát từ (derivative chromosome)
r: NST đóng vòng (ring chromosome)
i: Nhiễm sắc thể đều (isochromosome)
s: Vệ tinh (satellite)
t: Chuyển đoạn (translocation)
ace: Đoạn không tâm (acentric fragment)
add: Cộng thêm nhánh (vùng) của nhiễm sắc thể (additional)
cen: Phần tâm (centromere)
dic: Nhiễm sắc thể hai tâm (dicentric)
idic: Nhiễm sắc thể đều và hai tâm (isodicentric)
del: Mất đoạn (deletion)
dup: Lặp đoạn (duplication)
ins: Xen đoạn (insertion)
inv: Đảo đoạn (inversion)
rob: Chuyển đoạn hòa hợp tâm (Robertsonal)
rcp: Tương hỗ (Reciprocal)
mos: Thể khảm (mosaic)
Thể khảm (Mosaic)
Có
Không phân ly NST bất kỳ trong quá trình NP: hợp tử 2n nguyên phân rối loạn
Hiện tượng thất lạc NST: xảy ra trong NP
Rối loạn cấu trúc NST
- Là hậu quả của sự đứt gãy trên NST (gian kỳ). Nếu nối lại không đúng như cũ → Thay đổi cấu trúc NST.
Mất đoạn – del (Delections)
NST bị đứt rời 1 hoặc nhiều đoạn. 1/7.000
Đoạn bị đứt rời không có tâm → Tiêu biến.
Đoạn còn lại có tâm ngắn hơn NST tương đồng.
Có 46 NST trong tb, 1 NST ngắn hơn bình thường. → Mất chất liệu di truyền → Hậu quả nặng nề.
Có 2 dạng: Mất đoạn cuối và Mất đoạn giữa.
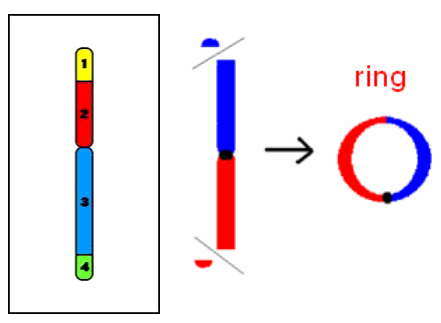
Mất đoạn cuối
Mất 1 đoạn ở phần cuối của NST: 46,XX,18p- hoặc 46,XX,del(18)(p21) hoặc 46,XX,del(18)(pter→p21:)
Mất cả 2 đoạn ở phần cuối của 2 nhánh NST: 46,XX,r(X)(p22q36) hoặc 46,XX,r(X)(::p22
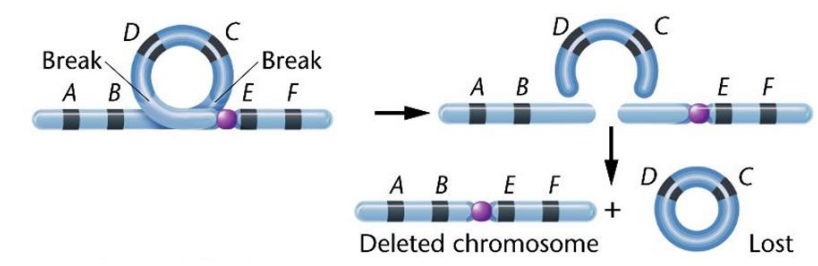
Mất đoạn giữa (Intercalary delection)
Sự uốn cong tạo thành vòng ở 1 nhánh của NST làm phát sinh 2 chỗ đứt gãy. Vòng không có tâm bị tiêu biến. Đoạn có tâm còn lại ngắn hơn bình thường.
Lặp đoạn – dup (Duplications)
Trên 1 NST, 1 đoạn được tăng lên 2 – 3 lần. → Tăng vật liệu di truyền.
Đặc điểm:
- Không gây hậu quả nặng nề như mất đoạn.
- Có lợi cho tiến hóa: Tạo chất liệu di truyền mới. Bảo vệ cơ thể không bị chết do mất đoạn.
46,XY,dup(1)(q22q25) hoặc 46,XY,dup(1)(pter
Có các dạng: Lặp nhánh p hoặc lặp nhánh q hoặc lặp cả NST.
Trên 1 cặp NST tương đồng:
- Có 3 nhánh ngắn → Trisomy p → 46,XY,dup(18)(p10)
- Có 3 nhánh dài → Trisomy q → 46,XX,dup(18)(q10)
LCRs (Low Copy Repeated sequences)
- Là các vùng khóa đặc biệt trên DNA, đặc thù ở mỗi NST
- Dài: 10 – 300 kb, thường ở gần tâm và 2 đầu mút.
- Chứa nhiều điểm dễ bị đứt gãy → NAHR → Mất (lặp) vi đoạn.
Mất vi đoạn – Lặp vi đoạn (< 5 Mb)
Sự tái tổ hợp tương đồng nhưng không tương xứng về alen (NAHR) giữa 2 chuỗi trình tự lặp lại sao chép thấp (LCRs) khác nhau trên 2 NST tương đồng ở GPI.
→ Hiện tượng mất (lặp) vi đoạn trên các NST này. Do lỗi tiếp hợp không thẳng hàng ở GPI (Misalignment)
→ TĐC không cân bằng (Unequal crossing-over).
LCRs: Low copy repeated sequences. NAHR: Non-allelic homologous recombination.
Nhiễm sắc thể đều - iso (isochromosome)
a. Nhân đôi 1 nhánh này và mất nhánh còn lại: 46,XX,i(12)(p10) hoặc 46,XX,i(12)(pter→cen→pter)
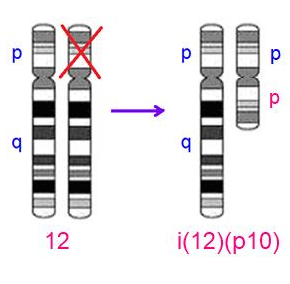
b. 2 nhánh nào đó được sao chép từ 1 nhánh nguyên gốc: 47,XX,i(12)(p10)
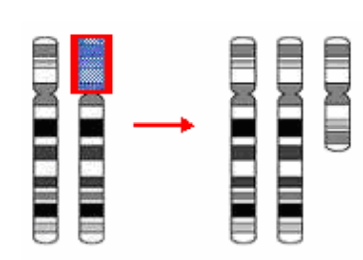
Nhiễm sắc thể 2 tâm – dic (Dicentric chromosome)
Hai NST cùng bị đứt:
- Các phần không tâm tiêu biến.
- Hai phần có tâm nối với nhau → NST 2 tâm (mất chất liệu di truyền, 45 NST). 45,XX,dic(8;8)(q14;q23) 45,XX,dic(5;10)(q22;q24)
Đảo đoạn - inv (Inversions)
Là sự sắp xếp lại NST.
Một đoạn NST bị đứt ở 2 chỗ và quay 180o. Hai chỗ bị đứt tự nối lại theo một trật tự mới.
Đảo đoạn ngoài tâm
Chiều dài và vị trí tâm NST vẫn như cũ.
46,XY,inv(1)(p12p31) 46,XY,inv(1)(pter→p12::p31→p12::p31→cen)
Đặc điểm:
- Không mất chất liệu di truyền. (nhuộm băng có thể xác định được)
- Làm thay đổi vị trí các gen.
Đảo đoạn quanh tâm
Mỗi nhánh đều bị đứt 1 chỗ. Hai chỗ đứt cách tâm không bàng nhau.
→ Vị trí tâm và chỉ số tâm thay đổi.
46,XY,inv(3)(p25q21) 46,XY,inv(3)(pter→p25::q21→p25::q21→qter)
Đặc điểm:
- Không mất chất liệu di truyền.
- Thay đổi trình tự các locus ở phần đảo đoạn.
Phân biệt đảo đoạn ngoài tâm và đảo đoạn quanh tâm
Dựa vào nhuộm băng
Chuyển đoạn – t (Translocations)
a. Chuyển đoạn tương hỗ (Reciprocal translocation hay Non-Robertson)
Trao đổi các đoạn giữa 2 NST không tương đồng. → 2 NST mới (khác nhau về hình thái nếu các đoạn trao đổi khác nhau về kích thước).
Đặc điểm: 46,XY,t(2;5)(q21;q31)
- Số lượng bộ NST không thay đổi.
- Có 2 NST mới bất thường.
- Chất liệu di truyền không thay đổi.
- Người mang 2 NST chuyển đoạn mới ở trạng thái cân bằng di truyền → Chưa thay đổi kiểu hình.
- Người này sinh ra con bị bệnh và thay đổi KH. Nhưng ?
46,XY,t(9;22)(q34;q11) 46,XX,t(9;22)(q34;q11)
- Bệnh Bc mạn dòng tủy; Ung thư máu dòng tủy mạn (Chronic Myelogenous Leukemia – CML)
- Là 1 q.trình ung thư chậm ở máu và tủy do đột biến chuyển đoạn tương hỗ giữa NST 9 và 22.
- Bệnh do đột biến mới phát sinh trong quá trình sống (nhiễm phóng xạ liều cao, kéo dài hoặc t.xúc hoá chất độc hại lâu ngày) → Không truyền bệnh cho đời con. Bệnh NST Philadelphia (Ph Chromosome) (Nowell & Hungerford, 1960)
abl: gen tiền ung (hoạt hóa Tyrosine kinase) kích thích tăng sinh tb.
Gen tiền ung (ABL) → Gen sinh ung (BCR-ABL) Làm tăng mạnh mẽ h.tính protein Tyrosine kinase. Tủy xương tạo dư thừa tb máu bất thường: Tb Myeloid (tb này chưa biệt hóa và không có chức năng). → Đi vào dòng máu, gọi là Blasts (nguyên bào). Khác với các b.cầu bình thường → Nhiễm trùng . Tủy xương: Blasts xâm lấn, giết các tb khỏe mạnh. (cũng không tạo ra hồng cầu, tiểu cầu khỏe mạnh).
b. Chuyển đoạn không tương hỗ (Insertions, ins)
Một đoạn của NST có thể:
- Chuyển đến NST khác nhưng không có ngược lại. ➔ NST này có thêm 1 đoạn và NST kia mất 1 đoạn. 46,XX,ins(5;2)(p14;q22q32)
- Hoặc chuyển đến vị trí khác cũng ở NST đó. ➔ Không làm ảnh hưởng NST. 46,XX,ins(2)(p13q21q31)
Chuyển đoạn cân bằng khi trao đổi chéo ở GPI tạo thành giao tử có chuyển đoạn không cân bằng
c. Chuyển đoạn hòa hợp tâm - Robertson
- Chỉ xảy ra ở các NST tâm đầu (D, G).
- Toàn bộ nhánh dài của NST này gắn lên toàn bộ nhánh dài của NST kia tại tâm. → 1 NST bất thường và 1 NST rất nhỏ bị tiêu biến.
Đặc điểm: 45,XX,rob(14;21)(q10;q10)
Chỉ có 45 NST ở kỳ giữa, đây vẫn là người lành, do:
- Thiếu 2 NST tâm đầu
- Xuất hiện 1 NST mới do chuyển đoạn → Kiểu hình vẫn bình thường (1/1.000)
Có tính di truyền hoặc do đột biến mới phát sinh
Khi kết hôn, người mang NST chuyển đoạn Roberson có nguy cơ mất cân bằng NST của các giao tử về sau. Hậu quả: Sẩy thai hoặc Đời con có thể bất thường
Vd 1: 1 NST 21 (nhóm G) với 1 NST 14 (nhóm D) Cấu trúc NST ở người lành có NST chuyển đoạn là: 14, 14/21, 21. Có 3 khả năng tạo các loại giao tử:
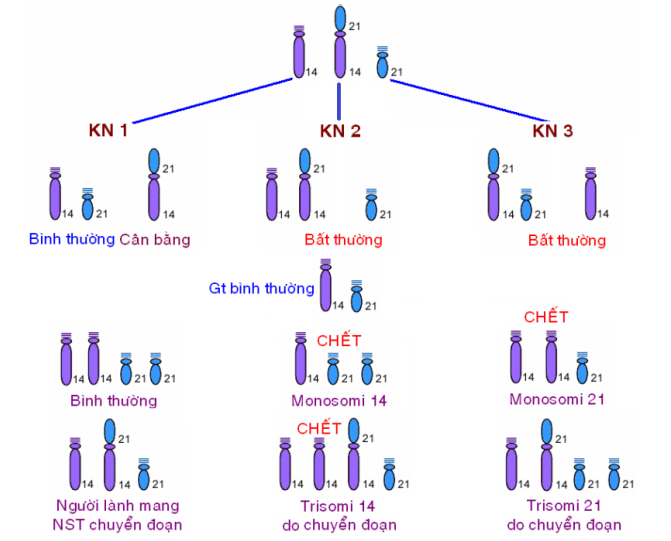
VD 2: 1 NST 21 chuyển đoạn với 1 NST 21 (G/G) Người mang NST chuyển đoạn (21/21):
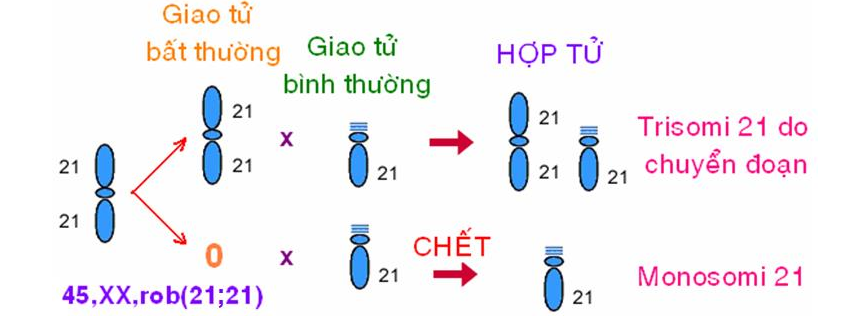
→ 50% người bệnh Down do chuyển đoạn (21/21). 46,XX,+21,rob(21;21)(q10;q10)
Dị Bội Thể (Aneuploidy)
Thiếu hoặc thừa một hoặc một vài NST ở một hoặc một số cặp NST trong bộ lưỡng bội.
Các dạng dị bội thể
- Thể không (Nullisomi: 2n – 2): Không gặp ở người
- Thể đơn (Monosomi: 2n – 1): Chỉ gặp trên NST giới tính ở người. Vd: 45,X
- Thể ba (Trisomi: 2n + 1): Đa dạng: 47,XXX ; 47,XXY ; 47,XYY 47,XY,+13; 47,XX,+18; 47,XX,+21
- Thể đa (Polysomi: 2n + 2; 2n + 3): Hiếm gặp ở người
- Thể Trisomi kép (Double trisomi: 2n + 1 + 1): Có thêm 2 chiếc NST ở 2 cặp NST bất kỳ.
Cơ chế gây dị bội thể
Bệnh liên quan đến NST thường
Mất đoạn
Hội chứng Cri du chat (5p-)
- 1/20.000
- 46,XX,del(5)(p15.2) hoặc 46,XX,del(5)(pter
- Nhẹ cân, dị dạng thanh quản, tiếng khóc như tiếng mèo
Hội chứng Jacobsen (11q-)
- 1/100.000 hiếm thấy
- 46,XX,del(11)(q24.1) hoặc 46,XX,del(11)(qter
- Khuôn mặt bất thường: Mắt xanh, Mũi nhô, Môi phồng
Hội chứng 18p-
Hội chứng 18q-
Hội chứng Ring 18
Lặp đoạn
Bệnh Trisomy:
- 47,XX,dup(18)(p10;q10)
Mất vi đoạn
H/c Wolf–Hirschhorn (4p-):
- (1/50.000-1/90.000)
- 46,XX,del(4)(p16.3)
- Trẻ nữ > nam 2:1
- Mất 1 vi đoạn
- trán và chóp mũi nhô lên, t’. Khoảng cách 2 mắt rộng và lồi hở miệng
Williams syndrome (7q11.23)
Miller-Dieker syndrome (17p13.3)
DiGeorge syndrome (22q11.2)
Angelman/Prader-Willi syndrome (15q11-q13)
Lặp vi đoạn
Hội chứng Charcot – Marie - Tooth: Lặp vi đoạn ở 17p12 (dài
Hội chứng Cat – eye: Lặp vi đoạn ở 22q11.2. (dài
NST đều
Hc Pallister-Killian Tetrasomy 12p (1/25.000) Có thêm một NST mới trong tb 47,XX,i(12)(p10)
DỊ BỘI THỂ
Trisomi 21 (hội chứng Down: DS)
1/800
Triệu chứng:
- Chiều cao bình thường.
- Thừa cân. Trán hẹp, gáy rộng, mắt xếch, dẹt, mũi ngắn và dẹt, tai nhỏ. Miệng thường mở, môi và lưỡi dày, hay thè lưỡi. Ngón chân cái tách ra.
- Trí tuệ kém phát triển, mặt dần độn, IQ thấp (<50).
NST 21 (225 gen) có vùng DSCR1 (Down Syndrome Critical Region 1) - 21q22.1
Tiến triển:
- 50% trẻ chết trước 5 tuổi.
- 8% trẻ sống trên 40 tuổi.
Nếu người nữ mắc h.c Down sinh con thì một số con cũng mắc Down. (vì 1/2 số giao tử của mẹ bị Down mang 2 NST 21)
Di truyền học tế bào
95% Trisomi 21 thuần: 47,XX,+21 ; 47,XY,+21 (88% do mẹ và 8% do bố)
Khảm: mos 47,XX,+21[2]/46,XX[4]
Downs do chuyển đoạn Robertson:
- 46,XX,+21,rob(14;21)(q10;q10)
- 46,XY,+21,rob(21;21)(q10;q10)
Thể ba 21 một phần (<1%): vùng DSCR1 (21q22.13)
- 46,XX,idic(21)(q22.3) + 46,XX,i(21)(q10)
- 46,XX,dup(21)(q22.1q22.3)
Trisomy 18 (Edwards syndrome)
1 / 7.500
Triệu chứng:
- Thường chết lúc bào thai. (do mẹ).
- Tuổi người mẹ cao thường ảnh hưởng đến đời con.
- Tỷ lệ: 4 trẻ nữ : 1 trẻ nam.
- Thai chậm phát triển và dừng ở tháng 7, suy thai
- Dị tật nặng: tim, thận và cơ quan s.dục
- Bàn tay nắm bất thường
Tiến triển:
- Sẩy thai, tử vong sau sinh.
- Rất xấu. Sống
Di truyền học tế bào (sống
- 95% Trisomi thuần: 47,XY,+18 ; 47,XX,+18
- 5% cơ thể khảm: mos 47,XY,+18[2]/46,XY[2] mos 47,XX,+18[1]/46,XX[2]
- Rất hiếm dạng chuyển đoạn hoặc trisomi kép 63
Trisomy 13 (Patau syndrome)
1/15.000
Triệu chứng:
- Đa số phôi thai bị sẩy.
- Tuổi mẹ có ảnh hưởng đến tần số con Trisomi 13.
- Sứt môi, hở hàm ếch.
- Dị tật nặng: TK, tim, thận, hệ tiêu hóa và bp s.dục.
- Rảnh khỉ, 6 ngón (thừa ngón út)
Tiến triển:
- 86% chết ở năm đầu. Rất hiếm 6 tuổi.
- 28% chết ở tuần đầu. 44% chết tháng đầu.
Di truyền học tế bào:
- 80% Trisomi thuần: 47,XY,+13 và 47,XX,+13
- 15% chuyển đoạn Robertson (13;14): 46,XX,+14,rob(13;14)(q10;q10)
- 5% Trisomi khảm: mos 47,XY,+13[2]/46,XY[1] mos 47,XX,+13[3]/46,XX[2]
Bệnh liên quan đến NST giới tính
Hội chứng Turner: 45,X
1/4.000
Triệu chứng:
- Sơ sinh: Thấp lùn, thừa da ở gáy.
- Dậy thì: Lùn, nhi tính.
- Ko kinh nguyệt. Tóc mọc ở gáy, cổ ngắn thừa da ở gáy, cẳng tay cong
- Ko có hormon sd, rối loạn dậy thì, vú lép
- Dùng Estrogens: Có dậy thì và
Di truyền học tế bào:
- 15% dạng NST X đều ở q: 46,X,i(X)(q10)
- Các dạng khảm: 5%: mos 45,X[2]/46,X,i(X)[q10](3) 15%: mos 45,X[1]/46,XX[2]
- 5% NST X vòng: 46,X,r(X)
- Các dạng hiếm (5-10%): 46,X,del(X)(p11.4) mos 45,X[3]/46,X,r[X](2)
Hội chứng siêu nữ XXX (Triplo - X)
Hay gặp hơn hội chứng Turner. 1/1.000 trẻ sơ sinh nữ.
5-10 trẻ sinh ra / ngày có XXX (Mỹ).
Triệu chứng:
- Không dị tật. Giới tính bình thường. Vô sinh
- Có thể vướng các vấn đề về tâm lí như lo âu, học kém, ảnh hưởng ngôn ngữ, kỹ năng vận động
- Ảnh hưởng dậy thì
- Như nữ bình thường. Không ai biết
Di truyền học tế bào:
- Dạng thuần: 47,XXX
- mos 47,XXX[2]/46,XX[3]
- Có 2 VT Barr (95% do mẹ)
- Hiếm: 48,XXXX; 49,XXXXX
Hội chứng Jacobs (Super-Male) 47,XYY
1/1.000 trẻ sơ sinh nam sống.
Triệu chứng:
- Cao và gầy. Hệ nội tiết bình thường. Testosterone có thể tăng cao.
- Lúc nhỏ: cao nhanh, gầy, mặt rỗ đầy mụn,...
- Lớn lên: giới tính bình thường. Có con.
- Như nam bình thường. Không ai biết
Hội chứng Klinefelter 47,XXY
1/1.000 trẻ sơ sinh nam (do mẹ)
Triệu chứng:
- Lúc sinh ra: không có dị tật và hình thái nam rõ rệt → Không chẩn đoán được.
- Lúc dậy thì: rối loạn cảm xúc, trí tuệ
- Dương vật nhỏ, tinh hoàn teo, mềm, không tinh trùng ➔ Vô sinh
- Ái nam ái nữ. Chân dài hơn thân.
Di truyền tế bào:
- Có 1 VT Barr
- 80%: 47,XXY
- 15%: mos 47,XXY[3]/46,XY[6]
- Hiếm gặp:
- mos 45,X[1]/47,XXY[3]/46,XY[2]
- mos 47,XXY[1]/46,XX[1]
- 48,XXXY; 48,XXYY; 49,XXXXY; 49,XXXYY
VẬT THỂ NHIỄM SẮC GIỚI TÍNH
Quan sát dưới KHV NST X, Y. Nếu thấy X, Y có trong: Tb đang phân chia NST GT Nhân tb ở gian kỳ Vật thể nhiễm sắc GT Có 3 loại:
DỊ NHIỄM SẮC CHẤT Có 2 loại dị nhiễm sắc chất:
- Dị nhiễm sắc chất ổn định. Vùng tâm và 2 đầu mút telomere → Cấu trúc vận động.
- Dị nhiễm sắc chất nhất thời. → Vật thể Barr
VẬT THỂ BARR
- Đa số tế bào ở nữ 46,XX đều có vật thể Barr.
- Xảy ra trong nhân tế bào soma ở gian kỳ (G1).
- Xuất xứ từ 1 trong 2 NST X bị ức chế, dị kết đặc và bất hoạt về di truyền (X bố hoặc X mẹ): Xi
Xảy ra ở nữ giới, phụ thuộc hoạt động sinh lý của: Tế bào và cơ thể.
Vật thể Barr – VT NS giới tính nữ
- Hình dạng: hình tháp, hình bán cầu, thấu kính lồi.
- Vị trí: nằm áp sát mặt trong của nhân tế bào.
- Bắt màu đậm so với nền nhân.
Vật thể Barr – Barr body
- Số lượng VT Barr = Σ NST X – 1 45,X 46,XY 47,XYY 46,XX 47,XXY 48,XXYY 47,XXX 48,XXXY 49,XXXYY 48,XXXX 49,XXXXY 49,XXXXX ➔ Chẩn đoán số NST X ở người bất kỳ
Đặc điểm vật thể Barr:
- Sự bất hoạt X: Ngẫu nhiên và không thuận nghịch (Xảy ra lúc phôi bào chứa khoảng 20 tb) → Giúp cân bằng di truyền: 46,XX và 46,XY
Gen SHOX (Xp22.33 và Yp11.3) PAR1 Mã hóa ra một yếu tố giúp điều hòa sự sao mã của các gen khác giúp xương phát triển. → Chiều cao + hình thái cơ thể cân đối.
Ứng dụng vật thể Barr Cơ chế hình thành mèo 2 màu và 3 màu:
- 2 màu (bi-colour): * Mèo Tortoiseshell (đen-vàng)
- 3 màu (tri-colour): Mèo Calico (đen-vàng-trắng)
Mèo tam thể / nhị thể Mèo cái: 38,XX. Mèo đực: 38,XY. Lông đen: gen b Lông vàng cam: gen O.
- Cả 2 gen này đều nằm trên NST X.
- Mèo ...... mang gen dị hợp tử Ob → Có 2 màu trên.
- Mèo tam thể gồm những mảng màu lông của bố xen lẫn với những mảng màu lông của mẹ.
- Mảng màu càng to (do chứa nhiều thế hệ tế bào con) khi X bất hoạt xảy ra càng sớm.
Hội chứng Lesch – Nyhan: bệnh di truyền lk trên X. (LNS: Lesch – Nyhan Syndrome) Do đột biến gen HPRT (Xq26) làm thiếu hụt E. HPRT. (HPRT: Hypoxanthine phosphoribosyltransferase) (giúp chuyển hóa Guanine + Hypoxanthine trong TĐC) Thừa Guanine và Hypoxanthine → Xanthine → A.Uric ➔ Bệnh Gout và sỏi thận.
Rối loạn cấu trúc NST X:
- 46,X,i(X)(q10) → VT Barr lớn hơn
- 46,X,i(X)(p10) → ? →
- 46,X,del(X)(p15) → VT Barr nhỏ hơn
❖ Các TH bất hoạt NST X không ngẫu nhiên: Nếu karyotype người bệnh có đột biến cấu trúc NST X dạng mất cân bằng (mất đoạn, lặp đoạn, NST đều) → X đột biến luôn được ưu tiên bất hoạt tạo VT Barr. → Tăng sức sống cho người bệnh
HỆ SINH DỤC NGƯỜI
Hệ sinh dục: gồm 3 phần:
- Tuyến s.dục: buồng trứng (nữ), tinh hoàn (nam).
- Đường s.dục: Nữ: vòi trứng, tủ cung, âm đạo, tuyến phụ thuộc, .... Nam: các ống dẫn tinh, tuyến tiền liệt, ....
- Bộ phận s.dục ngoài: Nữ: âm đạo, môi lớn, môi bé, âm vật. Nam: dương vật và bìu.
SỰ HÌNH THÀNH GIỚI TÍNH – HỆ SINH DỤC Qua 2 giai đoạn phát triển:
Gđ chưa biệt hóa. (Gđ trung tính)
Ống Muller (Ống cận trung thận)
Ống Wolff (Ống trung thận)
Gđ biệt hóa.
SỰ HÌNH THÀNH GIỚI TÍNH – HỆ SINH DỤC Gđ chưa biệt hóa (4 – 7w) Gđ biệt hóa (7w →) NST Y bất hoạt trong 6 tuần đầu của phôi thai. Không có testosteron, bpsd ngoài đi theo hướng nữ. Apoptosis
CÁC LOẠI GIỚI TÍNH Ở HỆ SD NGƯỜI
- Giới tính di truyền: 46,XX ; 50,XXXXXY: Nam
- Giới tính nguyên thủy: dựa trên loại tuyến sd. Tinh hoàn (nam); Buồng trứng (nữ).
- Giới tính nguyên phát: Đường sd + Bpsd ngoài. Nữ: tử cung, vòi trứng, âm đạo, môi lớn, bé, ... Nam: ống dẫn tinh, dương vật, bìu.
- Giới tính thứ phát: sau tuổi dậy thì (sinh sản). Nữ: có kinh, vú phát triển, dáng vẻ, giọng nói. Nam: có râu, dương vật và cơ bắp phát triển.
Khi không có NST Y: Hệ sd sơ khai → Buồng trứng → Hormon Estrogen → Âm đạo, tử cung, ống dẫn trứng (ống Muller ).
Khi có NST Y: Hệ sd sơ khai → Tinh hoàn → Hormon sd nam: AMH, Testosteron (ức chế đặc tính nữ) → Các đặc tính nam (từ ống Wolff ) → Cơ quan sinh dục nam. Hệ sd sơ khai không hoạt động → K.H nữ xuất hiện. Mọi người từ lúc sinh ra sẽ có K.H nữ trừ khi có xuất hiện các hormon sd nam.
NST GIỚI TÍNH Ở NGƯỜI
Nhiễm sắc thể X – Gen DAX1 (NR0B1)
NST X chứa > 150 triệu cặp base (> 1.400 gen). Gen DAX1 (Xp21.2) → 470 aa: kiểm soát tổng hợp các yếu tố quyết định: sự biệt hóa, sự trưởng thành và thực hiện các chức năng của buồng trứng. Các gen khác có liên quan trên X:
- Sự hình thành giới tính (b.trứng): WNT4 (1p35.1)
- Các tính trạng thường: SHOX (Xp), ...
Nhiễm sắc thể Y – Gen SRY
Bình thường: Nam 46,XY; Nữ: 46,XX Ngoại lệ (1/20.000): Nam: 46,XX Nữ: 46,XY.
- 1984, vùng TDF (Yp) quyết định tạo tinh hoàn. → Trẻ có Yp mà thiếu Yq sẽ là nam (ngược lại).
- 1990, gen SRY (223 aa) đặc trưng cho giới nam. SRY có ở: nam bình thường XY và nam XX. Không có ở nữ bình thường XX và bất hoạt ở nữ XY.
Sex-determining Region Y NST Y chứa: > 50 triệu cặp base (> 200 gen)
- Gen hình thành giới tính nam: Yp: SRY → Tinh hoàn (Yp11.3) SOX9 (17q24)
- Gen tạo tinh trùng ở Yq: AZFa, AZFb và AZFc (Yq11) Các rối loạn cấu trúc ở NST Y: 46,X,del(Y)(q21) 46,X,i(Y)(q10) 46,X,del(Y)(p14)
Tương tác giữa SRY và DAX1:
- SRY hiện diện, ức chế DAX1→ Tinh hoàn (nam).
- Thiếu SRY, DAX1 biểu hiện → Buồng trứng (nữ).
CÁC GEN TÁC ĐỘNG LÊN HỆ SINH DỤC 46,XX = 46,XY
Các yếu tố phát triển theo hướng nam
- TDF: quyết định tuyến sd sơ khai → Hướng tinh hoàn
- SRY: quyết định tạo tinh hoàn: tb Sertoli + tb Leydig. → Tiết ra AMH (MIF) và Testosteron.
- AMH: ức chế sự đường sd và bpsd ngoài của nữ.
- Testosteron → Đường sd và bpsd ngoài là nam. Nếu thiếu Testosteron: đ.sd và bpsd ngoài → Nữ hóa. (Mặc dù có hay không có Estrogen và Progesteron)
BỆNH LƯỠNG TÍNH
Người bệnh có tuyến sinh dục và bộ phận sd ngoài không đồng bộ. Phân loại dựa theo tuyến sinh dục:
- Lưỡng tính giả nam: có tinh hoàn (46,XY)
- Lưỡng tính giả nữ: có buồng trứng (46,XX)
- Lưỡng tính thật: có buồng trứng và tinh hoàn Các trường hợp lưỡng tính giả (thường gặp) có: Karyotype phù hợp với tuyến sd (trừ lưỡng tính thật)
Lưỡng tính giả nam
Đặc điểm chung: có tinh hoàn, 46,XY, VT Barr (–) Giới tính di truyền là nam, bpsd ngoài bị nữ hóa, do: Thiếu hụt Testosteron hoặc DHT DHT gây biệt hóa bpsd ngoài (tb kẽ tiết ra) Do có tinh hoàn nên tiết ra AMH: ống Muller tiêu biến Thường có dị tật lỗ tiểu dưới → Có tuyến sd và đường sd là nam Nhưng bộ phận sd ngoài bị nữ hóa.
Do thiếu enzyme 5 reductase-2 Enzyme này giúp chuyển Testosteron → DHT Do đột biến gen SRD5A2 (2p23) dạng p.Arg227Gln → Thiếu hụt enzyme 5 reductase-2 Nếu thiếu: không ảnh hưởng nữ nhưng ả.hưởng nam. → Dị tật lỗ tiểu dưới, nữ hóa bpsd nam: Bìu → Môi lớn Tinh hoàn còn nằm trong ống bẹn hay môi lớn. Vẫn có AMH: ống Muller tiêu biến, ống Wolff phát triển do có Testosteron → Có đường sd nam
Do thiếu enzyme 5 reductase-2 Khi dậy thì, lượng Testosteron tăng cao → Một số đặc điểm giới tính phụ: tăng lượng cơ, giọng trầm, lông mu nhiều, dương vật và bìu có lớn hơn, lông (râu) rất ít ở mặt và cơ thể. Người bệnh có tiên lượng tốt nếu được chẩn đoán sớm và phẫu thuật kịp thời < 2t để có con (hỗ trợ sinh sản). Khám và làm XN các enzyme, siêu âm đường sd. Hoặc: mổ thăm dò, sinh thiết tuyến sd.
Do thiếu Testosteron Đột biến gen 17 -HSD3 (9q22) làm thiếu hụt enzyme 17 hydroxysteroid dehydrogenase-3 Không có (rất ít) Testosteron → Ống Wolff không :
- Không có đường dẫn tinh.
- Tinh hoàn không di chuyển được xuống bìu.
- Bộ phận sinh dục ngoài bị nữ hóa. Do có AMH, ống Muller không phát triển. Khi dậy thì, Testosteron vẫn không có → Bpsd ngoài vẫn là nữ → Sự nữ hóa này không thể hồi phục (Ái nam ái nữ).
(do tăng CAG + GGC ở exon 1 ở gen AR) Gen tạo ra thụ quan Androgen (Androgen Receptor) bị đột biến → H.c thiếu hụt (kháng) Androgen (AIS). Androgen Insensitivity Syndrome: CAIS, PAIS. Tinh hoàn vẫn tiết testosteron nhưng thiếu hụt thụ quan Androgen → Testosteron không thể tác động mô đích. ➔ Hội chứng nữ hóa tinh hoàn 46,XY (H/c Morris) Xq11-12
Hội chứng nữ hóa tinh hoàn 46,XY VT Barr (–) Tinh hoàn ẩn. H.dáng, bpsd ngoài giống Nữ. (không có tử cung + buồng trứng) Tắt 1 đầu âm đạo, không có tử cung hoặc kém phát triển. Dậy thì: có vú, KH nữ, vô kinh, lông mu ít hay không có. Tâm s.lý hướng nữ, không biết bị bệnh, tự cho là nữ. Luật pháp + XH: luôn coi là nữ. Phôi thai học: giống lưỡng tính giả nam . → Mổ lấy tinh hoàn ngay khi phát hiện bệnh.
Hội chứng nữ hóa tinh hoàn 46,XY Rất hiếm, tỷ lệ 1/50.000 trẻ sinh ra sống. Được phát hiện khi đi khám Bs vì:
- Đến tuổi dậy thì vẫn vô kinh.
- Thoát vị bẹn (sa ruột). Hay: Tình cờ được khám xác định giới tính khi thi đấu thể thao. VĐV: Caster Semenya (1’56”) Chung kết 800m nữ ở Berlin 2009.
Lưỡng tính giả nữ (ít gặp hơn)
1/12.500. Có buồng trứng; 46,XX; VT Barr (+). ĐBG CYP21 (6p21.3) → Thiếu hụt enzyme steroid 21- hydroxylase → Cản trở corticoid tổng hợp bình thường → Androgen quá mức → Cường tuyến thượng thận bẩm sinh (nam hóa bpsd ngoài ở bé gái trước sinh). Nguyên nhân khác (hiếm), do: mẹ dùng androgen khi mang thai; mẹ bị u nang hóa buồng trứng tiết hormon nam hoặc tuyến thượng thận của bào thai bội tiết androgen (Tăng sản bẩm sinh → Có tính di truyền)
Lâm sàng: âm vật phì đại, môi lớn dính nhau. (biến dạng cấu trúc bpsd ngoài: nam hóa). Khi được điều trị thích hợp, bé gái có thể dậy thì bình thường và sinh con. Ngược lại, tình trạng nam hóa nặng tiếp tục đến trưởng thành (tạo 1 đoạn niệu đạo dương vật giống người nam có tinh hoàn ẩn).
Giới đảo nghịch (Sex Reversal)
1 / 20.000 80%: Nam 46,XX VT Barr (+). 1 / 50.000 20%: Nữ 46,XY ➔ Tuyến sinh dục khác hoàn toàn với karyotype (Bộ phận sinh dục ngoài phụ thuộc tuyến sinh dục)
- Rối loạn phát triển giới tính (DSD)
- Loạn sản tuyến sinh dục hoàn toàn (CGD) Có thể xảy ra trên NST giới tính hoặc NST thường. Nguyên nhân ??
Giới đảo nghịch trên NST giới tính
1966, Ferguson - Smith đưa ra giả thuyết phù hợp:
- 80% nam 46,XX: do chuyển đoạn SRY sang NST X.
- 20% nữ 46,XY: do đ.biến điểm hay mất đoạn SRY. Có SRY tạo hệ sd nam, không có sẽ tạo sd nữ. 10 – 15% nữ 46,XY do mất đoạn SRY. (46,XY,SRY–)
10 – 15% do đột biến điểm.
Bệnh sinh giới đảo nghịch Nam: 46,XX(SRY+)
GP tạo tinh trùng: TĐC vùng PAR1 giữa Xp và Yp. Bất thường do đột biến chuyển đoạn: Yp sang Xp mang theo gen SRY. Trên Yq còn có ít nhất 3 gen khác: AZFa, AZFb và AZFc tạo tinh trùng. Nếu thiếu dẫn đến vô sinh.
Kiểu hình và diễn biến Nam: 46,XX(SRY+ ) có các đặc điểm như TH: 47,XXY Thiểu sản tuyến sd, không tinh trùng, thoái hóa ống sinh tinh và vú nữ, vẫn có thể dậy thì tự nhiên. Dùng thêm Testosterone có đầy đủ giới tính thứ phát. Khác biệt: Nam 46,XX(SRY+ ) có vóc người bình thường, tỷ lệ xương và IQ bình thường, ít bị tâm thần. Nữ: 46,XY(SRY– ) cao bình thường, K.H như Turner, bị teo xơ tuyến sd nên không thể dậy thì tự nhiên.
Cơ sở di truyền Chẩn đoán xác định giới đảo nghịch phải dựa vào xét nghiệm FISH đối với gen SRY. Nam 46,XX(SRY+ ) và Nữ 46,XY(SRY– ) → Do tái tổ hợp mới.
- Bố mẹ ít có nguy cơ sinh con tiếp theo bị bệnh.
- Nếu bố mang chuyển đoạn cân bằng Xp với Yp, thì: Các con: Nam: 46,XX(SRY+ ) hoặc Nữ: 46,XY(SRY– ).
- Chúng đều bị vô sinh không truyền cho đời sau.
Giới đảo nghịch trên NST thường
- Gen SOX9 (17q24) → 509 aa. Có vai trò quan trọng trong các gen biệt hóa tinh hoàn. SOX9 thường xuất hiện với mức độ nhỏ không đáng kể theo sau sự biểu hiện của SRY thông qua SF1. SRY → SF1 → SOX9 → Tinh hoàn 46,XX: nếu lặp đoạn SOX9 → KH Nam (k.có SRY) H/c loạn sản Camptomelic: 46,XX,dup(17)(q23.1q24.3) WNT4, SF1, DAX1, SOX9 → Giới đảo nghịch Vd: Lặp DAX1 → KH nữ: 46,Y,dup(X)(p21.3p21.3)